阅读数:10870
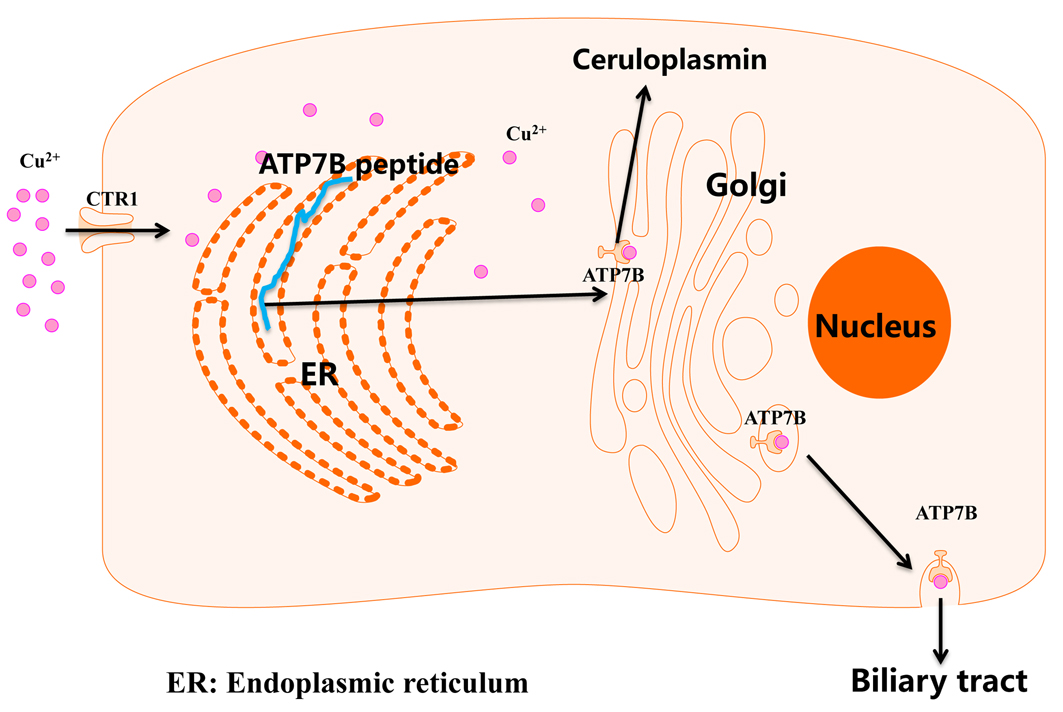
Wilson's disease is an autosomal recessive genetic disease, and ATP7B is the only known pathogenic gene responsible for it. ATP7B gene is mainly expressed in hepatocyte, and is localized to the Golgi membrane after being modified and processed by endoplasmic reticulum and Golgi apparatus. It is involved in the synthesis of ceruloplasmin and the excretion of copper through the biliary tract. When the body has excessive copper, Golgi apparatus transfers to the cell membrane in the form of vesicles, and then fuses with the cell membrane to discharge copper into the biliary tract.
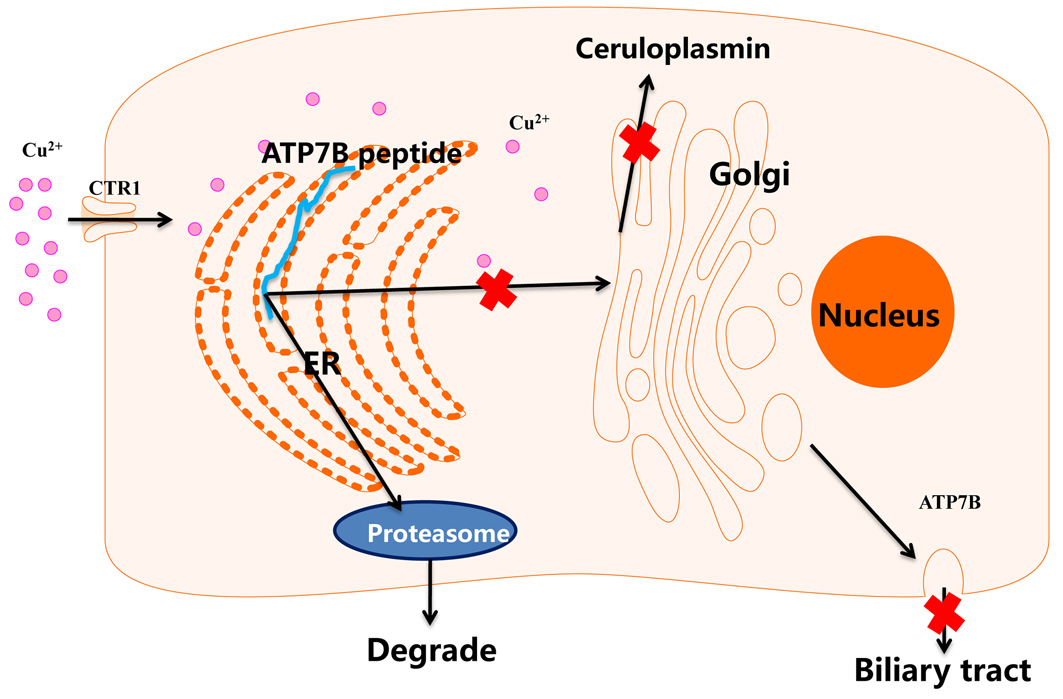
When ATP7B mutates, mutations in the 5'UTR region will affect the transcription of ATP7B. Nonsense, frameshift, and splicing sites mutations often cause ATP7B to translate a truncated ATP7B polypeptide, while missense mutations often cause protein folding, processing and cell positioning in a wrong path. The mutant proteins finally enter proteasome for degradation, and the ceruloplasmin synthesis and biliary copper excretion are impaired.
Reference:
1. Członkowska A, Litwin T, Dusek P, Ferenci P, Lutsenko S, Medici V, et al. Wilson disease. Nat Rev Dis Primers. 2018;4:21.
2. Chesi G, Hegde RN, Iacobacci S, Concilli M, Parashuraman S, Festa BP, et al. Identification of p38 MAPK and JNK as new targets for correction of Wilson disease-causing ATP7B mutants. Hepatology. 2016;63:1842–59.
3. Harada M, Sakisaka S, Terada K, Kimura R, Kawaguchi T, Koga H, et al. A mutation of the Wilson disease protein, ATP7B, is degraded in the proteasomes and forms protein aggregates. Gastroenterology. 2001;120:967–74.
4. van den Berghe PV, Stapelbroek JM, Krieger E, de Bie P, van de Graaf SF, de Groot RE, et al. Reduced expression of ATP7B affected by Wilson disease-causing mutations is rescued by pharmacological folding chaperones 4-phenylbutyrate and curcumin. Hepatology. 2009;50:1783–95.
